Bayesian phylogeographic inference of SARS-CoV-2
Nature Communications of 10 October has a paper by Philippe Lemey et al. (incl. Marc Suchard) on including travel history and removing sampling bias on the study of the virus spread. (Which I was asked to review for a CNRS COVID watch platform, Bibliovid.)
The data is made of curated genomes available in GISAID on March 10, that is, before lockdown even started in France. With (trustworthy?) travel history data for over 20% of the sampled patients. (And an unwelcome reminder that Hong Kong is part of China, at a time of repression and “mainlandisation” by the CCP.)
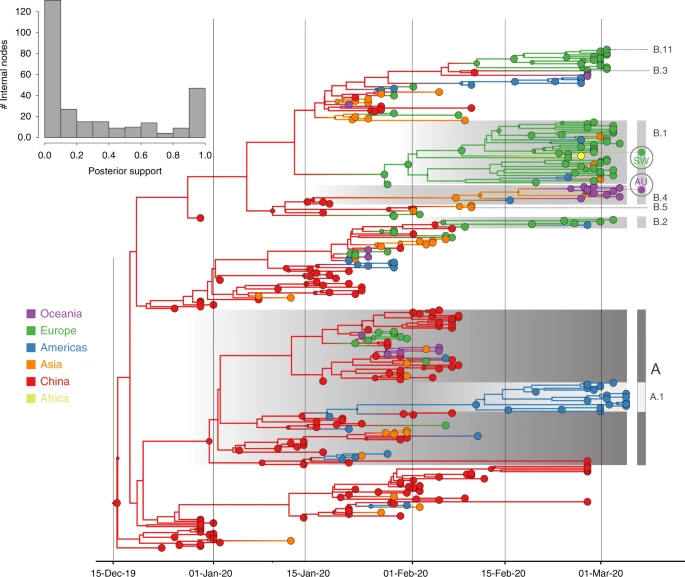
“we model a discrete diffusion process between 44 locations within China, including 13 provinces, one municipality (Beijing), and one special administrative area (Hong Kong). We fit a generalized linear model (GLM) parameterization of the discrete diffusion process…”
The diffusion is actually a continuous-time Markov process, with a phylogeny that incorporates nodes associated with location. The Bayesian analysis of the model is made by MCMC, since, contrary to ABC, the likelihood can be computed by Felsenstein’s pruning algorithm. The covariates are used to calibrate the Markov process transitions between locations. The paper also includes a posterior predictive accuracy assessment.
“…we generate Markov jump estimates of the transition histories that are averaged over the entire posterior in our Bayesian inference.”
In particular the paper describes “travel-aware reconstruction” analyses that track the spatial path followed by a virus until collection, as below. The top graph represents the posterior probability distribution of this path.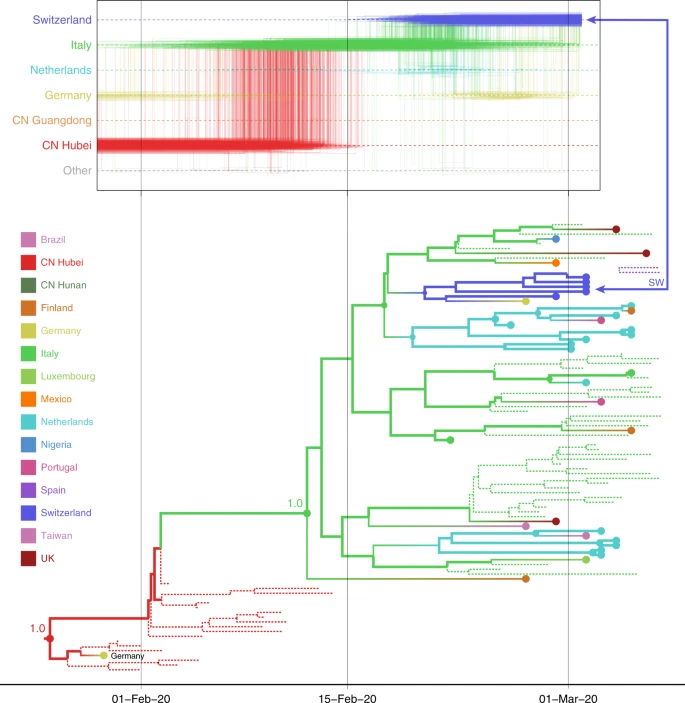 Given the lack of representativity, the authors also develop an additional “approach that adds unsampled taxa to assess the sensitivity of inferences to sampling bias”, although it mostly reflects the assumptions made in producing the artificial data. (With a possible connection with ABC?). If I understood correctly, they added 458 taxa for 14 locations,
Given the lack of representativity, the authors also develop an additional “approach that adds unsampled taxa to assess the sensitivity of inferences to sampling bias”, although it mostly reflects the assumptions made in producing the artificial data. (With a possible connection with ABC?). If I understood correctly, they added 458 taxa for 14 locations,
An interesting opening made in the conclusion about the scalability of the approach:
“With the large number of SARS-CoV-2 genomes now available, the question arises how scalable the incorporation of un-sampled taxa will be. For computationally expensive Bayesian inferences, the approach may need to go hand in hand with down-sampling procedures or more detailed examination of specific sub-lineages.”
In the end, I find it hard, as with other COVID-related papers I read, to check how much the limitations, errors, truncations, &tc., attached with the data at hand impact the validation of this philogeographic reconstruction, and how the model can help further than reconstructing histories of contamination at the (relatively) early stage.

Leave a comment